Nerves!
ø
I hadn’t quite anticipated this part—getting an interview for the proctor job has actually made me pretty nervous. Mateo came and picked me up at the house this morning for a jog to Harvard, so I finally broke the dry spell and ran for the first time in more than half a year. I am sore but happy about it. Mateo gave me a bit of a practice interview and a lot of (very helpful) pointers about what he thought they would probably be looking for. This is great, of course, but it did make me realize that I have quite a lot of preparation to do. They are undoubtedly going to ask me why I want to be a proctor, what the challenges are the students face, what my strengths are that I bring to the table, and other such predictable questions—and I’d better have well thought-out answers to all of those. Fortunately, I scheduled my interview for Wednesday morning, so the time I have to fuss over this is limited, and the time I have to get nervous before the interview on the day is brief.
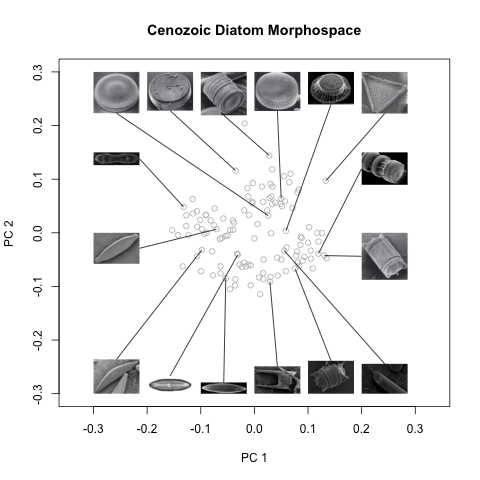
As hard as it is to tear myself away from obsessing over the preparations for this interview, I do need to get cracking on my morphospace. I started the morning by firing off emails to each of the authors of the most extensive diatom phylogenies I could find, asking them if they would share data that would enable me to construct one of the plots I outlined the other day (plotting morphological distance, i.e. pairwise dissimilarities from my d matrix, against genetic distances). Now it’s fingers crossed that this data exists, and if so, at least one of them is willing to share it with me.
In order to proceed, it might be helpful to re-sketch out a plan of action—as I’m getting that not-sure-where-to-start sensation, which is never good. Here’s what I think needs to happen:
- Implement Tinker’s suggestion of marginal totals for the PCO character loadings plot.
- Write the description for that plot.
- Figure out how characters are going to be referred to in the paper (X1-X123 of original matrix, or 1-74 of culled matrix).
- Make a table (?) of the x (where x is… 5? 10?) most important characters contributing to the first 3 PCO axes.
- Write a description of those most important characters.
- Choose a few (graphically, seven might be a good number) to represent around the edges of the PCO plot.
- Produce diagrams/drawings to represent those character states visually next to the subplots for those characters.
- Amend the drawShape() function to also reflect pattern center and raphe presence/absence (or find alternative solution).
- Try a 2*2D morphospace plot and see if it (axes 2 and 3, or 1 and 3) looks appreciably different.
- Construct the 2-D (or 2*2D) morphospace plot with the character plots and diagrams around the margin.
- Write a description of the morphospace plot and how it was made.
- Figure out how to get the topology of one of the diatom phylogenies (Sorhannus, Medlin, or Kooistra) coded so I can plot it in R.
- Select the genera from the tree that are also in my morphospace, and plot a phylogeny of just those in R.
- Color the taxa of the phylogeny by clade.
- Highlight the same colors on a 2D or 2*2D morphospace plot.
- Make a combined plot of phylogeny and PCO.
- Describe the phylogeny vs. PCO plot, how it was made, and what it shows.
- Finalize the morphospace through time plot.
- Obtain genetic data from Sorhannus, Medlin.
- Calculate genetic distances from sequence alignments.
- Compare results of different distance algorithms.
- Check and make sure the distance matrix from R matches that from MEGA (if it can be made to work).
- Plot genetic distance vs. morphological distance for Sorhannus data.
- Plot genetic distance vs. morphological distance for Medlin data.
- Describe the genetic distance plots and what it shows, and how it was made.
- Implement the bootstrapping algorithm that Foote used for his average pairwise distance calculations.
- Plot average pairwise distances—decide if for smaller time bins (per 2-myr?) or per stage.
- Find an algorithm for calculating alpha-shapes.
- Find a good alpha-value (it’s an arbitrary choice, right?).
- Calculate alpha shape volumes for each time bin.
- Repeat for convex hull volumes, which should be doable with the same algorithm.
- Implement SQ subsampling for diversity in R.
- Perform SQ subsampling on diatom data.
- Perform rarefaction subsampling on diatom data.
- Generate a big plot comparing various diversity measures with disparity measures.
- Write about how that plot was made.
- Write about what that plot shows.
- Go over the list of characters and pick some that might have biological meaning (linkage, defense, etc.)
- Make plots of absolute/relative abundance of those characters through time.
- Make big plot combining the trends in those characters through time.
- Describe what those plots show.
- Write code that will find the occurrence of characters (rather than genera) in a time bin.
- Sort the characters by first appearance.
- Make a plot of characters through time (range-through, presumably).
- Describe what that plot shows—highlight some of the more interesting first appearances and, if applicable, disappearances.
- Link the diversity rarefaction algorithms (see above) to the various disparity calculations (see above).
- Read up in Foote’s paper if he did anything else fancy with subsampling.
- Make a plot showing the effect of subsampling on the various disparity measures through time.
- Describe how the subsampling of disparity measures was done.
- Describe what the subsampling of disparity measures shows.
Phew! A little overwhelming… but it’s better to have it written out in a list I can check off item by item than having that shit all swirling around in my head.
Amazingly, in the course of the day, I got replies from both Sorhannus and Medlin, and Sorhannus had even sent his data by the evening. I spent Friday evening and most of Saturday working on the data, calculating genetic distances by various means and them correlating them with the morphological distances from my d matrix. By the end of Saturday I had a distance-vs-distance plot ready:

This is pretty interesting, because it’s not at all what I expected. I mean, I wasn’t expecting a perfect correlation or anything, but provided I haven’t fucked anything major up, there’s basically no correlation whatsoever between the morphological distances and the molecular distances. Wowzers! That either means morphological evolution is totally decoupled from molecular evolution (at least as measure by SSU rRNA sequences), or my morphospace is shit. Either way, an interesting result. I was also able to show that the choice of distance measure doesn’t have a huge effect on the strength of the correlation—the R-squared value is never much more than 0.05, is almost the same for most distance measures except for GG95 and TV, i.e. the Galtier and Gouy (1995) model where the G+C content may change through time and different rates are assumed for transitons and transversions, and TV, which is the number of transversions (if I decide to write that in the paper, I’ll have to understand what it means, first—I currently have no clue).

Well, I’d say it’s been a successful Saturday. I think I’ve accomplished something, both in getting a first result on the comparison of morphology to phylogeny, even though I don’t understand it, and in laying out a roadmap for the rest of the month. Even though it’s a long way to go, I now have a set of directions I can follow ‘blindly’ without having to worry too much at any time what my next task is supposed to be. Yay me. Now on to take a well-deserved few hours off.